



李六一医生的科普号
- 精选 进行性肌营养不良症-DMD的管理原则和策略
假肥大型肌营养不良症(DMD)是一种多见于男性儿童的肌肉疾病,是患病率很低的罕见病,其治疗方法和效果均无特异性,常用的有确切疗效的措施只是起到阶段性的效果或者效果甚微。但是患假肥大型肌营养不良症疾病的患儿家长,面对患儿的实际现状,应该在专科医生的指导下建立一个可持续性和有效果的应对措施,要有一个长期治疗的理念。每一个诊断该疾病的专科医生都有自己的治疗经验和相应管理措施。我做为一名神经内科医生,从事神经肌肉疾病诊断治疗是我的主要专业方向,针对DMD的诊断、治和预后谈谈自己的个人看法和相关措施。1.最关键的首先是明确诊断,即患者(患儿)是否是假肥大型进行性肌营养不良症或者Becker肌营养不良症,应该依据临床症状、家族史、以及起码的3项检测项目-肌酶谱、肌红蛋白和肌电图检查(有时候,患儿可能忍受不了针极肌电图);针对假肥大型肌营养不良症的明确诊断,以上各项资料和信息的获得,经一个负责任的医生综合考量肯定会得出一个确切的诊断结果。当然,为了获得更多的患儿和其家族成员是否是该疾病的基因携带者等状况,基因测序有时候也是必要的。但是针对患儿已经明确诊断是DMD,基因测序的意义有待商榷(费用太多)。2.患儿明确诊断为假肥大型肌营养不良症后,下一步就是治疗方法的选择和实施了。做为一名医生,我认为一定要与患儿的家属进行充分的交流和沟通,明确的告知他们:目前,国内外尚无对假肥大型肌营养不良症治疗方面的特殊措施,现在的治疗方法是暂时的缓解孩子临床症状的方案、或者是对症治疗和支持治疗,将来(1年或数年来)会有一个解决的办法的--不管是药物治疗、或是干细胞治疗和基因治疗(目前尚不成熟),假肥大性肌营养不良症患儿治疗方法的选择也应该让孩子家长能有效地参与其中。3.针对DMD患儿的治疗,我认为在孩子力量尚充足、但易跌倒的时候(5岁左右的年龄)就开始服用泼尼松片,服用方法依据患儿的年龄、体重和耐受程度来决定剂量、每天次数和持续时间(如果患儿肝脏有问题,可以换成美卓乐服用),同时需要服用保护胃部的药物、以及钙剂等药物,当然服用泼尼松片也应该在适当的时候进行一些调整和剂量的改变。另外的药物包括:ATP,辅酶Q10、谷维素片,肌苷片,肌生注射液,对于DMD患儿也有帮助;非假肥大型肌营养不良症患者服用强的松是没有可靠疗效的。4.中药或中成药对于DMD是否有作用呢?有些医生会认为中药不起什么作用。但是,就我本人近几年来治疗患儿经验来看,对DMD患儿(或BMD)的用药方面:黄芪片、补中益气丸等药物对于部分患病孩子的肌力提升、耐受运动程度提高还是具有一定裨益的。因此,对于DMD患儿的治疗措施多角度多方位的考量,应该不失为一项有意义的尝试和探索。5.使用支具、心理关爱和有效的护理、营养疗法等措施也会大大地给予患者帮助和解决某一个方面问题的,这些方法尽量在临床医生的指导下,在适当的时候让患者积极的使用。6.至于患儿治疗过程中泼尼松片的用法、何时停药均应在主管医生的指导下进行,如果该撤药时没有及时撤掉,不仅没有益处反而有不利的地方;别的口服药物也应在医生指导下按照患儿的病情、症状、病程、体重、年龄和对药物的反应等等状况来具体进行的即个体化用药措施,我最反对一些医生不顾患儿的具体情况、症状,而一味的、毫不改变的长期用药治疗。7.至于基因疗法、干细胞治疗等最新的医学治疗技术目前尚不成熟应用于临床应用于患儿,但是未来必将会在治疗DMD疾病方面有很大的价值。也一定会有确实的效果的;至于新的治疗DMD药物的研发和应用,已经在欧美国家已见雏形,肯定在不长的时间里会应用于治疗DMD患者的临床中。8.至于跳跃51等经国外批准使用的药物,进入国内的时间是指日可待的,但是也需要一段时间才能应用于临床和造福于患者。9.以上针对假肥大型肌营养不良症患儿诊断治疗的思考,是我做为医生在诊疗DMD患儿过程中的一些经验和建议,仅是个人观点,肯定有不妥之处,仅供参考,敬请多多指正!本文系李六一医生授权好大夫在线(www.haodf.com)发布,未经授权请勿转载。
李六一 主任医师 河南省人民医院 神经内科103人已购买 - 精选 重症肌无力患者(MG)日常生活中需要注意的事项和管理
MG疾病是一种慢性疾病,无论是服用最好的药物治疗还是采取最合适的手术治疗,都不是立竿见影就会效果大显的情况,从患病开始到临床治愈结束,需要有一个时间过程,即病程较长,因此MG患者在这个治疗疾病的过程中(3-4年)需要注意以下几点事项: 1.治疗疾病需要有打持久战的准备,要有信心,要保持一个好的轻松的心情来接受这种疾病治疗的现有状况。即MG患者心理上要有一个充分的接纳丶接受的治疗疾病的适应过程和了解状态;同时要有信心,MG是一种可以治疗的疾病,在正确合适的治疗原则下会逐渐康复的。 2.避免剧烈运动和耐受度强的活动,包括肢体和脑力两个方面。依据患者自身的身体素质和健康状况,进行适量的运动和采取健康的活动方式,始终保持一个好的体质,锻炼的"度"一定要把握清楚,可以采取多次短时间活动方式,譬如散步和安全水域的游泳丶原地的肢体关节伸展舒张等活动,运动时间尽量选择在自己状态好的时候,达到自我感觉"微累"即停止持续运动。 3.在专科医生指导下,持续服用药物-包括药物品种丶剂量丶用法和调整药物等,尽量不要私自减少剂量和突然停药,和在服用药物过程中自己私下增加药物品种,在调整药物丶换药和改变药物方法等事情上,都应该与自己的主治医生及时反馈和沟通。 4.在治疗疾病的过程中,尽量避免自己在公共场合的次数,防止呼吸道感染和感冒,在天气变化之际根据冷热状况及时更换衣服(增加或者减衣服),最好在治疗期间养成一个好的饮食起居(例如易食用易消化的食物,少食生冷酸辣和坚硬的食物)习惯,防止胃肠道丶呼吸系统发生疾病和感染,从而继发MG疾病出现症状危重丶加重等不良后果。 5.由于治疗MG的药物譬如溴呲斯的明经常短缺,建议在购买药物时,尽量备足后2个月治疗的药物用量;同时,要定时复诊,评估MG病情进展状况,及时调整相关药品;至于MG的手术治疗方法,需要经专科医生进行仔细的评估后,而且年龄适合,药物治疗效果差,合并有胸腺增生或者有胸腺瘤者可以实施手术治疗。 6.MG患者治疗期间,时间跨度长,避免不了外科手术丶妊娠丶失眠和焦虑抑郁等情况,麻醉方式及麻醉用药丶和胎儿发育和生产丶对应药物的选择这些必要的事情,都应当在专科医生临床评估和安全性评估后再进行,可能更合适。 7.在长期的治疗疾病过程中,要基本了解服用药物治疗或手术治疗的一些副作用,初步知道可能一些症状是新疾病的临床表现还是由于治疗MG的不良反应,及时反映给自己的主治医生,有一个基本的应对措施。 8.不要偏信"偏方密方治疗重症肌无力,一药见效",这均是骗人的把戏,如果相信了这些骗子的手段,只能毁了自己的身体和耗费了自己的血汗钱,不仅会让自己后悔莫及,也肯定会导致重症肌无力危象。 本文系李六一医生授权好大夫在线(www.haodf.com)发布,未经授权请勿转载。
李六一 主任医师 河南省人民医院 神经内科309人已购买 - 精选 简述“运动神经元病”(渐冻症)的必要知识,渐冻人-是什么
一:运动神经元病受累的范围1.脊髓前角细胞2.脑干运动神经核3.皮层锥体细胞和锥体束。二:运动神经元病的发病状况发病率:年发病率约2/10万,人群患病率4-7/10万,90%以上为散发病例,预计我国国内该病的患者有六万余人(河南省可能有5000余患者)成人患病情况:通常在40~60岁起病,男性多见,可能与男性的雄性激素调节环节有关。遗传性:5%的患病者有遗传性,其中有20%可以检出为SOD1基因突变。三:运动神经元病的病因1.遗传因素:SOD1基因突变是运动神经元病发病机制中研究最多的内容之一,仅仅占5%以下的比率。SOD1是一种人体所有细胞内高表达蛋白,称为铜/锌超氧化物歧化酶。2.兴奋性氨基酸毒性。3.自由基氧化损伤。4.神经细丝及神经元变性。5.线粒体形态和功能异常。6.环境因素和病毒感染。运动神经元病是一种异质性疾病,许多的研究所涉及的假说都不能解释MND发病的整个过程,可能只是运动神经元病发病的一个阶段或者环节。究竟是什么原因何种机制导致了运动神经元损伤的过程,以及MND仅仅运动神经元选择性受累,并没有一个明确的结论。四:运动神经元病的临床分型1.肌萎缩侧索硬化症(ALS,就是网络上常说的“渐冻症”疾病,患者经常被称为“渐冻人”)2.进行性脊肌萎缩(PSMA)3.进行性延髓麻痹(PBP),也称球麻痹4.原发性侧索硬化(PLS)五:辅助检查1.神经电生理检查:四肢肌电图检测是诊断和鉴别诊断“渐冻症”患者最重要的评估项目。1)肌电图和神经传导速度测定:是诊断运动神经元病最有价值的手段,特别是在疾病早期,尤其具有诊断和鉴别诊断的价值。2)运动诱发电位(锥体束检查,MEP):此项检查为MND提供上运动神经元受累的客观依据,因为ALS和PLS均有中枢运动传导时间延长。3)单纤维肌电图检查(SFEMG)2.MRI检查3.脑脊液检查。六:运动神经元病的诊断要点1.中年隐袭起病,慢性进行性发展病程。2.肌无力、肌萎缩、肌束震颤并腱反射亢进、病理征等上、下运动神经元受累征象。3.神经电生理检查显示在4个节段(脑干、颈、胸、腰骶髓4个节段),至少3个节段存在神经源性损害。4.无感觉障碍,无尿便障碍。5.排除可以解释临床表现的其他疾病。七:运动神经元病的疾病鉴别非典型运动神经元病病例必须与以下疾病鉴别1.脊肌萎缩症(SMA)2.脊髓型合并神经根型颈椎病3.多灶性运动神经病(MMN)4.单肢肌萎缩(平山病)5.ALS叠加综合征6.X-连锁脊球神经元神经病(Kenney病)7.脊髓空洞症8.脊髓灰质炎后综合征9.颈椎合并腰椎神经根病10.CIDP11.良性肌束震颤八:运动神经元病的治疗MND的治疗尚无有效方法。MND的治疗目的是通过各种有效的方法和切实的措施:改善患者生存质量和生活质量,延长患者的生命。1.利鲁唑,又名力如太,是目前唯一被美国FDA批准用于治疗ALS(运动神经元病,渐冻人)的药物,成人剂量为50mg,2次/天,服用时需要定时进行实验室检测。2.各种维生素类药,大剂量口服,效果因人而异。3.中药治疗(有些验方及成药能给予患者一些帮助)。4.对症治疗。5.支持治疗和心理治疗。6.综合治疗和有效的护理方法。
李六一 主任医师 河南省人民医院 神经内科1.5万人已读 - 精选 腓骨肌萎缩症的简述
腓骨肌萎缩症(perineal muscular atrophy),又称为Charcot--Marie--Tooth病(CMT),该病由法国的Charcot和Marie以及英国的Tooth等三位医学专家在1886年首先进行描述的,CMT又称遗传性运动感觉性周围神经病(HMSN),是最常见的遗传性周围神经病的一种类型,其特点如下:1、有家族史,多见于常染色体显性遗传。2、发病年龄:儿童期或青少年期发病,发病率男性为女性的4倍左右。3、临床表现:进行性四肢远端肌无力和肌萎缩,多伴有“仙鹤腿”及“弓形足”,有感觉减退症状,腱反射减弱或消失,肢体多呈对称性受累和缓慢进展的病程。4、据神经电生理结果和病理特征可将CMT分为2型:CMT1型----脱髓鞘型;CMT2型----轴索型。5、周围神经活检有“葱球样改变”。6、该病预后尚好,许多患者发病后仍可生活数十年,积极治疗和对症治疗可以提高病人的生活质量。本文系李六一医生授权好大夫在线(www.haodf.com)发布,未经授权请勿转载。
李六一 主任医师 河南省人民医院 神经内科5977人已读 - 知识普及 眼咽型肌营养不良,需与重症肌无力、ALS(渐冻症)进行鉴别!
【OPMD,容易与重症肌无力、运动神经元病(渐冻症)等疾病混淆的一种肌肉疾病】1.眼咽型肌营养不良(OPMD)介绍:OPMD呈常染色体显性遗传。特点:发病年龄晚,临床上以进行性加重的眼睑肌下垂、吞咽困难和四肢无力为特征,病情以进行性加重为主。2.OPMD的临床诊断标准:1)、一般50岁左右发病;2)、临床表现为进行性加重的睑下垂、吞咽困难、发音障碍;3)、查体可见眼肌、咽部肌的无力和萎缩;4)、血清CK正常或轻度升高;5)、肌电图检查为肌源性改变;6)、肌肉活检为肌源性损害;7)、有家族遗传病史,呈常染色体显性遗传;8)、病情进行性加重。3.分析与建议:1)、在临床上遇到具有睑下垂、吞咽困难和发音障碍的患者的情况下,一定要详细地询问患者的病史,并且做必要的实验室检查项目,区分三种不一样的神经系统疾病;2)、尤其是神经肌电图检查项目,针对这三种临床症状相似却是不同的疾病的时候,行肌电图检测的数据和结果是根本不同的,即肌电图检查结论有三种不同的表现,其可以对应以上三种不同的疾病,为临床上明确诊断、预后和治疗方案选择提供精准的客观信息。(图片大部分来自网络上)
李六一 主任医师 河南省人民医院 神经内科363人已读 - 典型病例 只有一个患者初步诊断为:渐冻症!
周三上午看门诊有三个患者在家里人的陪同下,说自己是"渐冻症"这个疾病。其中有一个年龄只有19岁的患者,才是一个严重的"疑病症者",有20天的全身肌肉跳动症状史,自己认为自己就是渐冻症,已经在省内找过6个医院的7个医生就诊过,找我的时候让我诊查右手部第一指骨背间肌肌肉酸困无力的次数达5次之多,最后终于以正常结束。三个自己或家属认为是运动神经元病的患者中,这其中只有这一个患者经考量患者的起病状况、临床症状和体征、病程进展、辅助检查结果等综合判断为:肌萎缩侧索硬化症(ALS,渐冻症)。这个患者的情况见下图图片所示:患者,男,34岁。主诉:四肢无力,肌肉跳动1年半,近期症状明显加重,同时伴有说话不清、肌肉萎缩加重。你说这个患者是不是渐冻症呢?!
 李六一 主任医师 河南省人民医院 神经内科2541人已读
李六一 主任医师 河南省人民医院 神经内科2541人已读 - 知识普及 【"我是不是渐冻症呀?!" 2例严重疑病者的就诊记录】
实际上,门诊就诊的这2例患者都是年轻人,第1例患者(图片示)是22岁、男性,是在校大学生;第2例患者,女,35岁,可能从事文化教育工作。 一、第一例患者(图片所示),是在其父亲的陪同下进入诊室的,走路的时候,好像下肢有点拖曳无力的样子,小伙子个子挺高,也挺帅的,但是满面的愁容,抱着一种无奈的心态,坐在诊椅上。 "小伙子,怎么不舒服?" "医生,我是不是渐冻症呀!?" 噢,我有点猛一楞的感觉,看看年轻人的面容。 "你怎么知道自己是渐冻症呢?" "下肢的肌肉跳动,手上肌肉也跳动,我上网络查询一下,就看到渐冻症了,吓死我了" 该患者面部表情有许多焦虑,他的父亲也是直直地站在那里双眼紧张地盯着我,也是有着一股纠结的心情。 我先安抚了患者的情绪,让他的父亲也拉个诊椅坐下。 问了他的病程和病史、目前的症状。 患者讲了他的情况:一个月前因打蓝球比赛,不慎摔倒导致右脚踝受伤,实际上并没有骨折只是皮外伤或轻度肌腱损伤,经及时处理治疗后好转,但行走时还有些右下肢运动受限、有些力弱,在家里休息养伤的时候,上手机上查询自己的症状,看到了一些渐冻症(ALS)疾病的信息:早期ALS患者症状不对称,有一个肢体无力表现加重,并逐渐有肌肉跳动的症状等,随即就感觉自己的右下肢有肌肉跳动,几天后左下肢也有,双上肢手部也开始无力了同时也有肉跳,该患者就认为自己是:渐冻症,恐惧、纠结和痛苦随即发生,他的父亲听了他的讲述也吓得不轻,及时到当地医院神经科就诊,没有什么大的问题,依然不放心,全家人都很焦虑和紧张,就至省医院就诊来解决这个问题。 这的确是一个大问题! 我很快问询了该患者的一些病情并做了体格检查:从双下肢、上肢和颅神经等均发现异常体征,只是右踝部外侧局限区域有点色素沉着(可能是皮外伤留下的)。 初步诊断排除运动神经元病,可能是躯体化症状,焦虑状态。 患者的面部有了笑容,他的父亲也轻松了许多。 实际上不需要做什么检查项目,服用一些药物调整一下心态和自己做些调适情绪即可。 但该患者还是强烈要求做四肢肌电图检查,我就给他开了EMG检查单,安排科里医生做了检查,结果是正常的。 肌电图结果出来后,我又与该患者和其父亲一起做了沟通,并简介地解读ALS(渐冻症)疾病的一些基本知识,并说明了为什么他不是渐冻症这个疾病。 父子俩个非常高兴地离开了诊室,小伙子面带微笑不停地说谢谢。 疑病症,的确害人不浅! 二、第二例,实际上是一个复诊的患者,她与2020年、2021年曾经因肢体无力、肌肉跳动等症状,怀疑自己是肌萎缩侧索硬化(ALS,渐冻症),经诊查和数次肌电图详细检测结果,已经排除渐冻症的可能。 此次该患者因单位做体育活动项目,用力不平衡引起了下肢的不适感觉,休息几天后仍然未见好转,就又一次怀疑可能是ALS(渐冻症)早期。 患者走进诊室后,我就认出了她,问她有怎么不适的症状呢,她就说下肢无力好多天没有见好,是不是渐冻症呢? 我依据患者的情况,又一次给她讲解了关于渐冻症的部分知识,以及不支持她是渐冻症疾病的原因。 当然,又是一个自己要求做肌电图检查的人,实际上需要嘛?! 结果肯定是正常的。 针对该二位怀疑自己是"渐冻症疾病"的患者,启发如下: 1.有临床症状和不适的体征的情况下,到底自己是什么疾病?是不是是很严重的某些罕见病例如ALS(渐冻症),不要盲目地上网络上拿自己的症状去比对具体疾病,然后猜测自己或者亲属是什么疾病,这不仅于事无补,而且会害了自己,也会给自己的家里人带来一段时间内的痛苦。 疾病是什么或不是什么,这个诊断和鉴别诊断结论应该让专科医生来处理和确认; 2.实际上在这个医患关系有点微妙的时代里,我认为许多医生还是都站在患者的角度来考虑疾病这个事情的,相信医生和相信医生的诊疗过程,做为患者来说,对于明确诊断疾病是非常必要的; 3.针对疾病方面,现代诊疗技术水平的发展也有许多不可知的区域也有很多变化的因素,也有局限性,但毎个医生都是在努力着的。
 李六一 主任医师 河南省人民医院 神经内科2177人已读
李六一 主任医师 河南省人民医院 神经内科2177人已读 - 知识普及 运动单位电位(MUP)是肌电图检查的关键收集之一
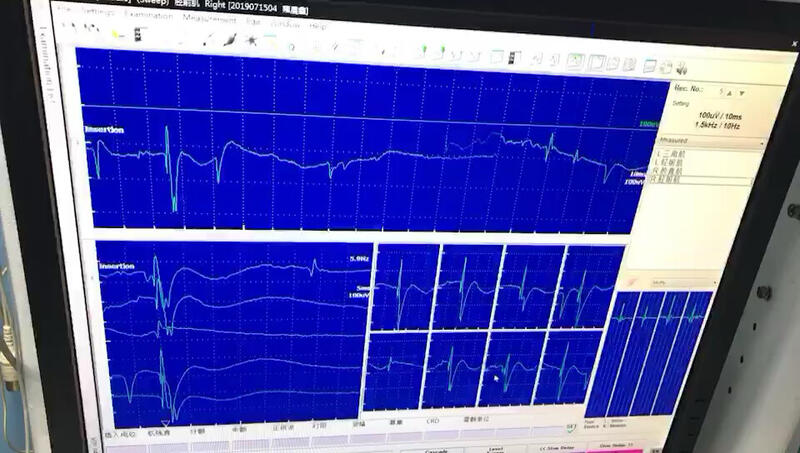 李六一 主任医师 河南省人民医院 神经内科1612人已观看
李六一 主任医师 河南省人民医院 神经内科1612人已观看 - 知识普及 阅读分享:渐冻症介绍
概述:MND以脊髓前角细胞和选择性脑运动神经核团变性为特征; 是罕见病; 1.肌萎缩侧索硬化(ALS,这个疾病实际上的俗称是"渐冻症"),发病率1-2/100000,是最声名狼籍的运动神经元病,ALS在身体上还是精神上都是最具破坏力的神经系统疾病之一; 2.分类:ALS(肌萎缩侧索硬化)、PMA(进行性肌萎缩)、PBP(进行性延髓麻痹)、原发性侧索硬化(PLS); 3:预测MND患者个体的病情进程和最终结局具体在什么时候是困难的; 一些患者迅速进展为ALS(或本身就是ALS)预后会很差; 一些患若则持续表现为局限性MND是更为延长的结果,预后尚可; 4.MMN(多灶性运动神经病)不是MND疾病,它是一种显著的可治疗疾病,诊断MMN主要依据肌电图结论和抗GM1抗体; 5.病因学:尚不明确,研究支持MND的产生影响因素有:神经微丝结构和功能障碍、线粒体损害、谷氨酸兴奋毒性、自由基毒性与氧化损伤、小胶质细胞激活的神经再生损害等,但尚未确定具体的疾病机制,近期有学者研究发现MND的发病机制可能与C9ORF72基因发生突变有关联。 6.ALS诊断中的问题 ALS的诊断基于其典型的临床发展过程,并为辅助检查结论所提供依据; 特异性的确定一个患者是否是MND疾病,而且如果是,会是何种MND?是困难的,尤其是早期,MND/ALS的诊断不确定性是令人困惑的,许多时候也会让医生处于怀疑的矛盾之中。 如果患者起病有运动神经损害的临床表现,并伴有进展性UMN和LMN功能障碍,而且病情发展范围超过最初累及的解剖区域,并排除了潜在的其他神经系疾病,又有辅助检查(肌电图)结论的证据,MND/ALS的临床诊断会更明确了。 MND/ALS疾病一般认为早期病程呈线性发展,但针对不同的个体患者在发展速度变化上可相差30倍; 评估方法如肌力测定和用力肺活量的测量,其结论对于个体患者预后情况如何是可靠的指标。 7.诊断方法:肌电图(EMG)、脑和脊髓的影像学检查、肌肉活检及血液生化检查等是常用的手段; 基因测序; 经颅磁电刺激(MEP)、PET、MRl波谱检查也可以为明确诊断提供依据。 8.MND的治疗 利鲁唑 依达拉奉等药物; 症状性治疗即对症施治:虽然不能满足患者的要求和医生的期望,但它可使患者在舒适状态、功能和安全性得到一些实质的改善;另外还能提高患者和医生对MND的信心; 支持治疗:可以改善患者的舒适程度、功能提升和安全性。 图片1:著名理论物理学家 史蒂夫·霍金,渐冻症患者; 图片2:世界首富 比尔·盖茨,为渐冻症患者募集基金做"冰桶试验"。 理论物理学家 霍金 渐冻症患者 比尔·盖茨 冰桶挑战试验
 李六一 主任医师 河南省人民医院 神经内科1694人已读
李六一 主任医师 河南省人民医院 神经内科1694人已读 - 知识普及 肌肉代谢的能量来源与运动不耐受性肌病
一、肌肉代谢所需的能量来源依赖于循环的葡萄糖和游离脂肪酸。 安静状态下,肌肉利用游离脂肪酸作为代谢的能量需求; 在低强度运动的过程,葡萄糖优先做为能量来源,脂肪酸在长期运动后由脂质氧化动员而来; 在高强度运动的时候,代谢的能量来源主要是储存于肌肉的糖原氧化酵解最重要,剧烈运动时转变为无氧酵解; 以上肌肉的代谢过程中每一个节点出现异常都会导致肌肉疾病发生。 二、运动不耐受性肌病 大致来说共有3种: 1.糖原累积病 磷酸酶缺乏(McArdle病) 磷酸化酶激酶缺乏 磷酸果糖激酶缺乏 磷酸甘油酸脂激酶缺乏 磷酸甘油酸变位酶缺乏 乳酸脱氢酶缺乏 2.呼吸链缺陷疾病 复合物1缺乏 辅酶Q10缺乏 复合物3缺乏 复合物IV缺乏 3.脂质代谢障碍疾病 肉毒碱棕榈酰基转移酶2缺乏 长链脂酰辅酶A脱氢酶缺乏 短链3羟酰辅酶A脱氢酶缺乏 三功能蛋白缺乏
 李六一 主任医师 河南省人民医院 神经内科3216人已读
李六一 主任医师 河南省人民医院 神经内科3216人已读

李六一主任医师
河南省人民医院神经内科
